RAMTools: Extending ROOT for Genomic Data Processing

Introduction
Hello! I’m Aditya Pandey, and this summer I had the privilege of participating in Google Summer of Code (GSoC) 2025 with CERN-HSF as part of the Compiler Research Group. It has been an incredible experience working with my mentors, Vassil Vassilev and Martin Vassilev, on a project that bridges the gap between high-energy physics (HEP) and genomics.
Project Overview
RAMTools is a project that extends ROOT—CERN’s data processing framework—to efficiently handle genomic sequencing data. While ROOT was designed for petabyte-scale physics data, its cutting-edge features are perfectly suited for the challenges of modern genomics.
The core problem with traditional genomic formats like SAM/BAM is that they are row-oriented, making analytical queries on massive datasets slow and inefficient. My project introduces RAM (ROOT Alignment/Map), a new system that leverages ROOT’s latest columnar format, RNTuple. This provides:
- Columnar Storage: Optimal for fast analytical queries and high compression ratios.
- Parallel I/O: Built-in support for concurrent read/write operations.
- Modern Compression: Support for multiple algorithms (LZ4, LZMA, ZLIB, ZSTD).
By converting SAM data to the RNTuple format, we can achieve significant performance gains in both storage and query speed.
Technical Implementation
The project was implemented in C++17 and built using CMake, relying on the ROOT framework (version 6.26+) for its RNTuple I/O subsystem.
Architecture Components
-
SAM Parser: A custom, high-performance C++17 parser optimized for streaming and processing extremely large SAM files.
-
RNTuple Writer: An efficient data model that maps the fields of a SAM record (QNAME, FLAG, RNAME, POS, etc.) to a columnar RNTuple structure.
-
Chromosome Splitter: A key feature that allows for partitioning the output into separate files by chromosome, enabling trivial parallel processing of downstream analysis.
-
Region Query Engine: A fast query tool that leverages RNTuple’s selective column reading to extract genomic regions (e.g., chr1:10150-10300) without reading the entire file.
Command-Line Tools
The primary interaction with RAMTools is through two command-line executables:
SAM to RAM Conversion (samtoramntuple)
Converts a standard SAM file into the optimized RNTuple-based RAM format.
# Basic conversion
./tools/samtoramntuple input.sam output.root
# Split by chromosome for parallel processing
# (Creates output-chr1.root, output-chr2.root, etc.)
./tools/samtoramntuple input.sam output -split
Region Querying (ramntupleview)
Queries a specific genomic region from a RAM file, similar to samtools view.
# Usage: ./tools/ramntupleview [input.root] "[chromosome]:[start]-[end]"
./tools/ramntupleview output.root "chr1:10150-10300"
Performance Achievements
We benchmarked RAMTools using the HG00154 sample from the 1000 Genomes Project, which consists of 196 million reads in a 72.1 GB uncompressed SAM file.
Query Performance Comparison
RNTuple’s columnar architecture shows significant speedups, especially for large region queries, when compared to the older ROOT TTree format and CRAM (industry-standard compressed format).
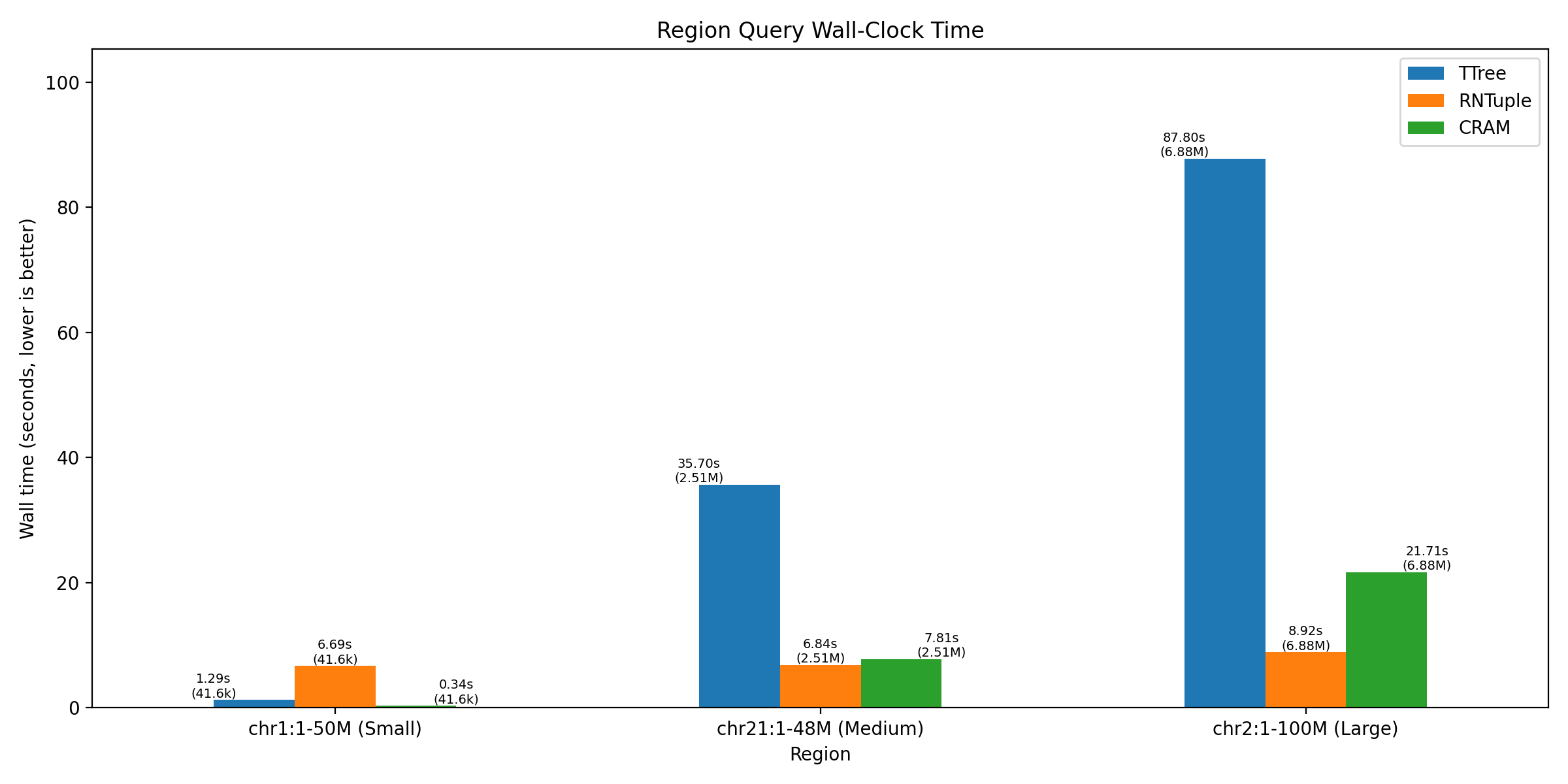
The benchmarks demonstrate performance across three query sizes:
| Query Region | Size Category | Region Coordinates | RNTuple Time (s) | TTree Time (s) | CRAM Time (s) |
|---|---|---|---|---|---|
| Small | 50M | chr1:1-50M | 6.69 | 1.29 | 0.34 |
| Medium | 48M | chr21:1-48M | 6.84 | 35.70 | 7.81 |
| Large | 100M | chr2:1-100M | 8.92 | 87.80 | 21.71 |
For the small region (chr1:1-50M), CRAM performs best due to its reference-based compression optimizations for sequential access. However, as query size increases:
- Medium queries (chr21:1-48M): RNTuple is 5.2x faster than TTree and competitive with CRAM
- Large queries (chr2:1-100M): RNTuple is 9.8x faster than TTree and 2.4x faster than CRAM
The performance advantage of RNTuple becomes more pronounced with larger analytical queries, making it ideal for whole-chromosome or multi-gene region analyses common in genomics research.
Storage and Compression
RNTuple also provides excellent compression. The 72.1 GB SAM file was compressed down to 11.4 GB using ZSTD, a 6.3x compression ratio.
| Format | Compression Algo | File Size (GB) | Additional Requirements | Total Storage (GB) |
|---|---|---|---|---|
| SAM | Uncompressed | 72.1 | - | 72.1 |
| CRAM | Reference-based | 7.8 | 3.2 GB reference file | 11.0 |
| RAM-RNTuple | ZSTD | 11.4 | Self-contained | 11.4 |
| RAM-TTree | LZMA | 12.5 | - | 12.5 |
| RAM-TTree | ZLIB | 16.7 | - | 16.7 |
| RAM-TTree | LZ4 | 31.2 | - | 31.2 |
The most significant achievement here is that the 11.4 GB RNTuple file is completely self-contained. This is a key advantage over formats like CRAM, which achieves a similar total storage size (11.0 GB) but is dependent on an external 3.2 GB reference genome. This self-contained nature simplifies data archival, distribution, and use in cloud environments immensely.
Repository & Documentation
- GitHub: RAMTools Repository
Future Work
While GSoC has concluded, there is a clear path forward for RAMTools:
-
More format Support: Support for more formats for wide adaptation.
-
Further Query Optimization: Explore multi-threading in the query engine to parallelize data retrieval.
-
Integration with Analysis Frameworks: Investigate integration with popular bioinformatics frameworks or visualization tools.
Conclusion
GSoC 2025 has been a phenomenal experience. I’ve had the opportunity to dive deep into high-performance C++ and solve real-world problems in genomics.
I am immensely grateful to my mentors, Vassil Vassilev and Martin Vassilev, for their invaluable guidance, insightful code reviews, and constant support. I also want to extend my thanks to the entire ROOT team, CERN-HSF, and Google for making this project possible. I look forward to continuing my contributions to this exciting intersection of science and technology.

